使用命令提交并行运行作业
本章节以运行 Lammps 软件应用程序为例,详细介绍在 EHPC 集群中通过命令行提交作业、查看作业结果的具体流程,从而使用户可以更好的理解 EHPC 集群中命令行的基本使用逻辑。
作业文件说明
Lammps 软件应用过程中,需要用到的作业文件包括:
-
计算文件,文件格式为
*in或.lj。 -
其他相关文件,文件格式为
.restart,__airebo,__lcbop等。
在本次示例中需要用到 .lj 计算文件和执行脚本文件 .sh,用户可在附录中查看或下载源码。
操作步骤
-
说明 本示例中所创建的 EHPC 集群登录节点和管控节点采用的默认配置,计算节点则使用 4 台 2 核 4G 的通用计算节点。
-
等待集群创建完成后,将 Lammps 作业所需的文件(
.lj和.sh文件)上传至 EHPC 集群的共享目录中,详细步骤可参考上传文件相关章节。说明 在本示例中需上传的文件为附录中的 ball.lj 和 ball.sh 文件。
-
参考 WebSSH 章节相关内容,登录至集群的登录节点。
-
依次执行如下
module命令,加载 OpenMPI, Lammps 以及 IntelMPI 模块。说明 若想要了解更多
module相关命令,可执行module --help。-
加载 OpenMPI 模块。
module load openmpi -
加载 Lammps 模块,版本为 20190430。
module load lammps/20190430说明 -
若不指定版本号,此命令会默认加载相应模块的最新版本,若需加载其他版本,可执行
module avail lammps命令查看当前系统可支持的模块版本后,再执行module load lammps/20190430命令加载指定版本,其中20190430为模块版本号,需根据实际情况进行修改。 -
执行
module unload lammps,可卸载相应的模块,其中 lammps 为模块名称需根据实际情况进行修改。
-
-
加载 IntelMPI 模块,此处需加载 intel/18.0. 1 版本,与 lammps/20190430 相匹配。
module load intel/18.0.1
-
-
执行如下命令,查看上述模块是否加载成功。返回结果中会显示当前系统中已加载的模块文件。
module list回显示例:
Currently Loaded Modulefiles: 1) hpcmodulefiles 2) lammps/20190430 3) openmpi/4.1.1 4) intel/18.0.1 -
执行如下命令,进入上传有 Lammps 作业相关文件的目录下,即步骤 2 文件上传的路径。
由于本示例中,运行所需文件被上传至共享目录下新建的
/test目录: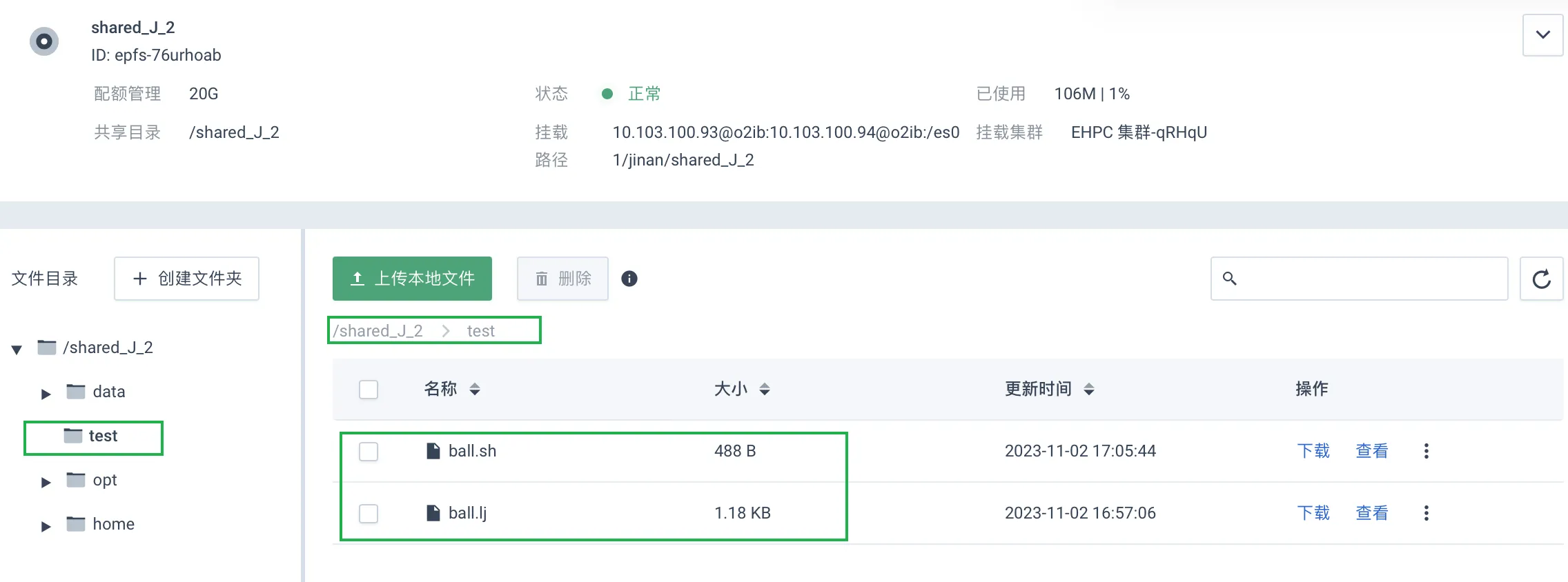
故,此处执行命令如下:
cd /es01/jinan/shared_J_2/test查看当前目录下的所有文件。其中 .lj 为 Lammps 的计算文件,.sh 文件为作业运行脚本,其详细内容请参看附录
ls -alh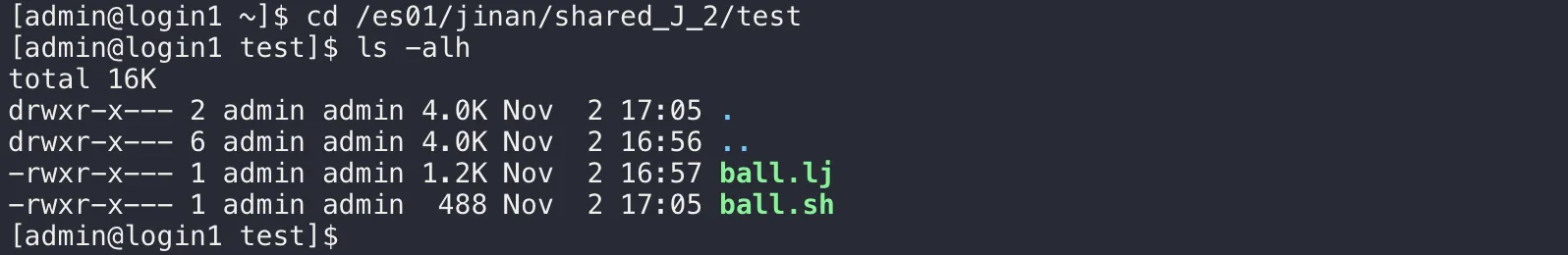
-
执行如下命令,提交作业。作业提交成功后,会显示相应的
job id,如下所示本示例的 job id 为3。sbatch ball.sh回显示例:
Submitted batch job 3 -
在弹性高性能计算 EHPC 控制平台页面,选择左侧导航栏中的作业管理,上述步骤中提交的作业已显示在列。
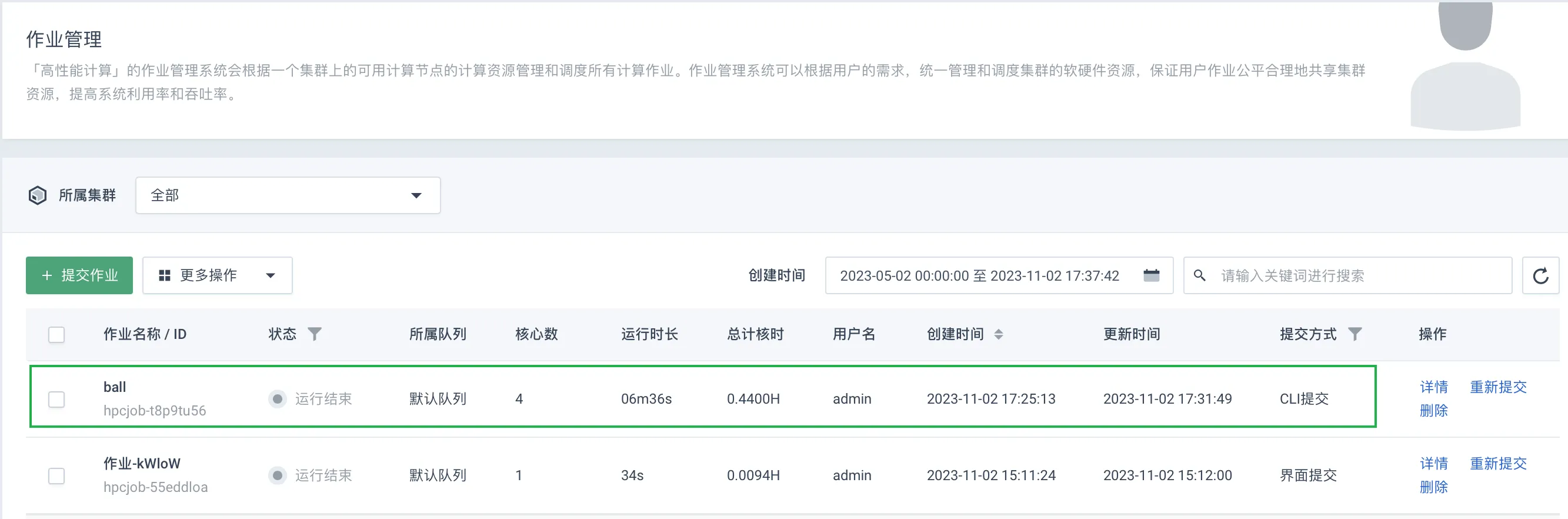
-
点击相应作业操作列中的详情,进入其详细信息页面,选择计算目录,选择计算文件上传的路径,本示例为
/test文件夹,用户可自主查看或下载指定文件。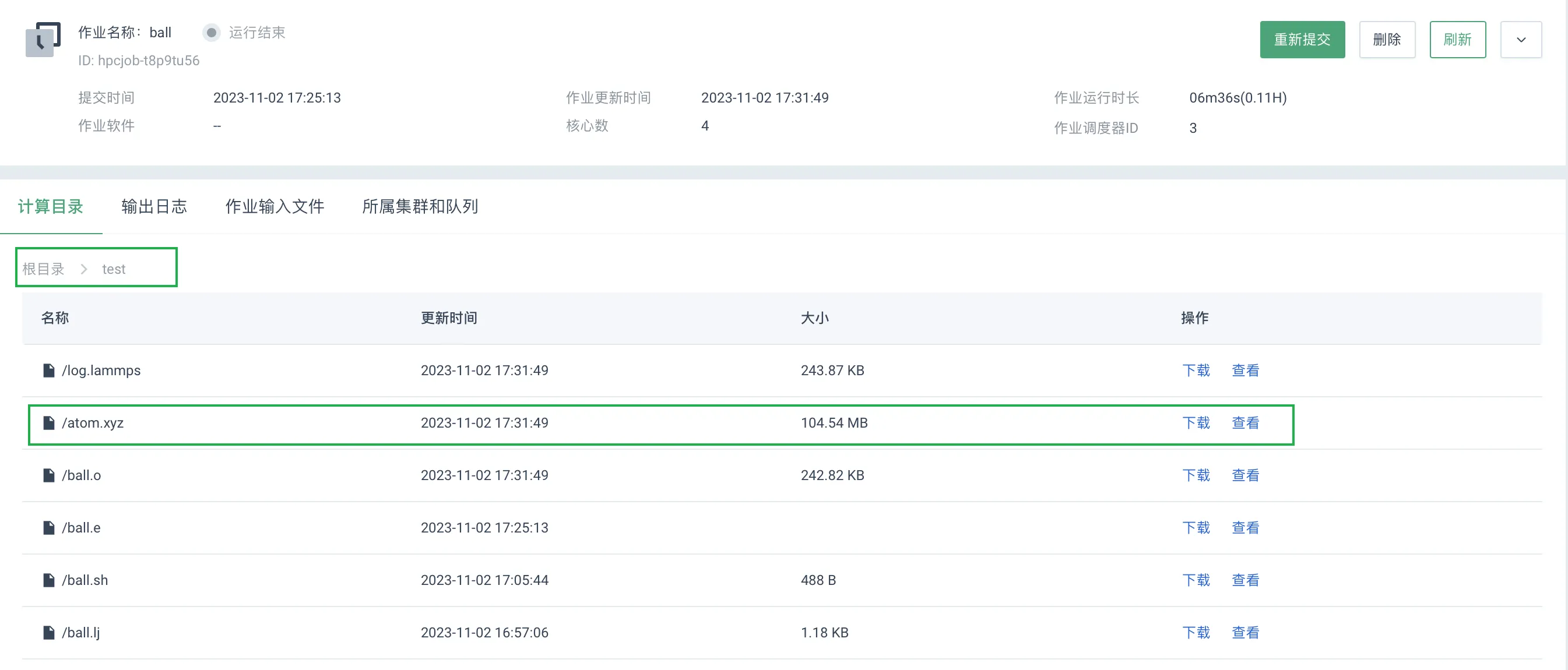
-
将
atom.xyz 文件下载至本地后,上传至 VMD 软件查看结果即可。
附录
计算文件
本次示例中使用到的 Lammps 作业的计算文件源码 ball.lj 内容如下,可从此处下载。
#原子数量
variable npart equal 800
#体系单位lj
units lj
#二维体系
dimension 3
#原子类型atomic
atom_style atomic
#周期性边界
boundary p p p
#近邻参数
neighbor 6 bin
#邻居列表更新频率
neigh_modify every 1 delay 0 check yes
# box 尺寸
region box block -20 20 -20 20 -20 20
#在 box 内生成 2 种原子
create_box 2 box
#转为二维计算
#fix 3d all enforce3d
#在 box 内随机生成 800 个原子,原子类型为1
create_atoms 1 random ${npart} 324523 box
#随机生成 1 个类型为 2 的原子
create_atoms 2 random 1 32524523 box
#原子质量
mass 1 1
mass 2 5
#设置力场参数, soft 势
pair_style soft 1.0
pair_coeff 1 1 10.0 1.0
pair_coeff 1 2 10.0 3.0
pair_coeff 2 2 10.0 3.0
#温度初始化
velocity all create 2.0 34234123 dist gaussian
#能量最小化,防止原子重叠
minimize 1e-4 1e-4 1000 1000
#步数初始化为0
reset_timestep 0
#输出设置
#dump img all atom 10000 atom.xyz
# nve 系综
fix integrator all nve
#热力学输出
thermo_style custom step temp ke pe
thermo 100
#模拟步长
timestep 0.001
#启动模拟
#输出设置
dump DUMPFILE all xyz 100 atom.xyz
run 500000执行脚本
执行脚本 ball.sh 内容如下,也可从此处下载。
#!/bin/bash
#SBATCH -J ball # 指定任务名称
#SBATCH -n 4 # 指定核心数量
#SBATCH -N 4 # 指定 node 的数量
#SBATCH -p normal # 提交到哪一个分区
#SBATCH --mem=2000 # 所有核心可以使用的内存池大小, MB 为单位
#SBATCH -o ball.o # 把输出结果 STDOUT 保存在哪一个文件
#SBATCH -e ball.e # 把报错结果 STDERR 保存在哪一个文件
#SBATCH -t 1-5:00 # 运行总时间,天数-小时数-分钟, D-HH:MM
mpirun -np 4 lmp_mpi -in ball.lj